Research
Areas of Research
Myopathic diseases are a substantial economic burden for long-term care; while symptoms themselves may be treated there are currently no cures available. Our research program is focused on the identification and characterization of proteins essential in muscle development and maintenance with the ultimate goal of bridging the knowledge gap between basic research and new targets for therapeutic interventions. Simpler genetic models, such as Drosophila melanogaster, have been instrumental in uncovering new genes that are conserved in higher organisms to unravel the cellular basis for protein dysfunction. Our laboratory uses genetic, biochemical, and cellular biology approaches to understand the basic molecular events that underlie muscle formation and homeostasis and how defects in normal muscle function may lead to the onset and progression of myopathies. To achieve this overall goal, we are pursuing multiple avenues of study: (1) understand how the muscle-tendon interface, or myotendinous junction (MTJ) is formed and maintained; (2) examine mechanisms of muscle homeostasis and how misregulation of these processes may result in disease; and (3) unravel the role of Dock family members in morphogenesis of the Drosophila heart tube.
Protein Aggregation
Non-dividing muscle and nerve cells have limited options to clear harmful biological insults. One such insult is the progressive accumulation of damaged and misfolded proteins that ultimately destroy cellular function and may result in cell or organismal death. Understanding how and why normal protein turnover occurs in healthy tissue is essential for the eventual treatment of age-related and/or degenerative diseases that result from abnormal protein accumulation. Using the large, easily manipulated muscles in fruit fly larvae, we find that loss of the evolutionarily conserved NUAK protein results in cellular degeneration due to the abnormal accumulation of certain proteins, including Fil and CryAB. Moreover, we have identified Stv/BAG3 and its binding partner Hsc70-4 to be crucial for NUAK function. Current experiments are aimed at identifying the triggers and cellular machinery that are required to clear protein aggregates with the long-term goal of uncovering potential therapeutic targets for the treatment of protein aggregate myopathies.
Drosophila as a Model System for MTJ Biogenesis and Stability
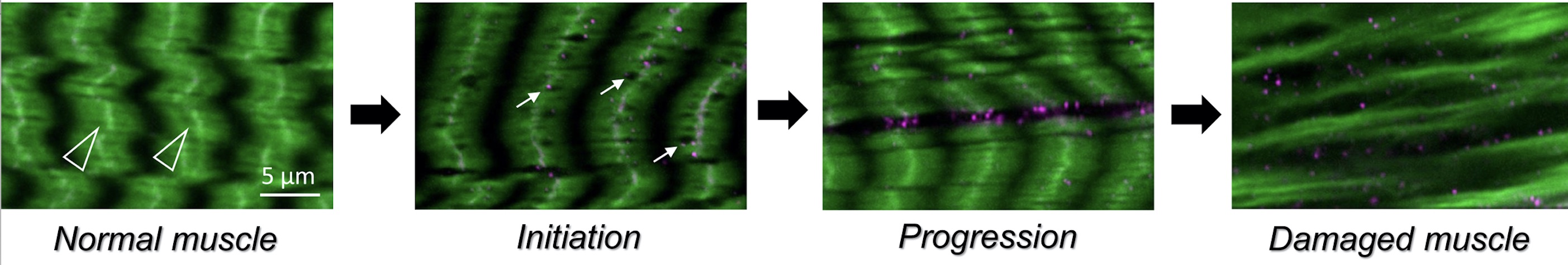
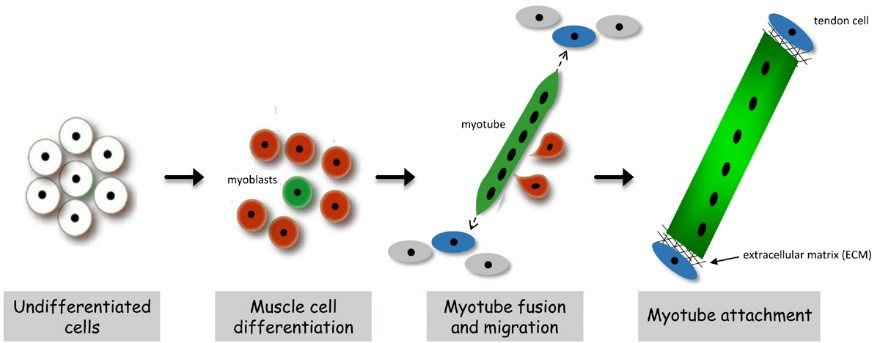
In both invertebrates and vertebrates, muscle development proceeds through an exquisitely regulated series of myoblast fusion and myotube guidance events that allow muscles to grow in size and form stable MTJs. The MTJ is a highly-specialized adhesion site between muscles and their corresponding tendon cells. Structurally, the MTJ is comprised of secreted extracellular matrix (ECM) proteins that are physically tethered to integrin complexes expressed on the plasma membranes of both muscle and tendon cells. In vertebrates, the tendon is the connective tissue that transmits forces created in the muscle to the bones. Invertebrate model organisms, such as Drosophila, lack an internal skeleton but serve as an excellent model to study the conserved processes of MTJ formation and maintenance, as Drosophila has pioneered the identification of genes required for all phases of myogenesis. In flies, the contractile muscles are firmly attached to specialized types of epidermal cells, called tendon cells, which anchor the muscles to the external cuticle, or exoskeleton. In all organisms that possess contractile muscles, muscle attachment defects may lead to immobility and lethality. Thus, the formation and maintenance of the MTJ requires tight regulation of cellular processes including gene expression, intracellular protein transport, and protein turnover.
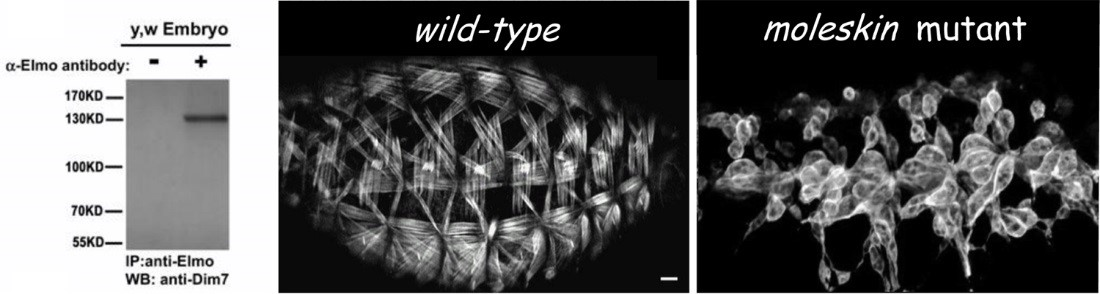
MTJ Formation and Stability
Instability of the MTJ is one underlying cause of congenital muscular dystrophies (CMD), and the progressive muscle weakness resulting from these myopathies is an obvious detriment to human health. The MTJ is the primary site for force transmission from the interior of the muscle cell, across its membrane, and to the extracellular matrix (ECM). In healthy muscle tissue, the MTJ provides resistance against mechanical stress generated during muscle contraction. It is now known that any decrease in MTJ stability leads to muscle detachment in diverse organisms, including humans. Our work initially focused on unraveling the role of the Engulfment and Cell Motility protein (Elmo) in myogenesis. In a series of genetic studies, we showed that Elmo functions in concert with Myoblast city (Mbc)/Dock180 as an unconventional guanine nucleotide exchange factor to activate the small GTPase Rac in myoblast fusion. Subsequent examination of additional elmo alleles highlighted a new role for Elmo in muscle attachment. This finding provided us with an entry point to uncover new players required for embryonic MTJ formation. We chose to deviate from traditional genetic screens and utilized a protein-based approach, capitalizing upon the identification of Elmo as a previously unrecognized component that localizes to the MTJ. To this end, tissue-specific immunoprecipitations (IPs) were carried out to identify proteins that interact with Elmo in the developing muscle. The protein with the highest peptide coverage that emerged from these studies was Mbc, which nicely validated our previous Elmo-Mbc biochemical and genetic interaction data. Implementation of this in vivo proteomics screen resulted in the identification of additional Elmo-binding proteins, namely Drosophila Importin-7 (DIM-7), encoded by the moleskin (msk) gene. Mutations in msk are lethal and defective in muscle attachment. While the canonical role for Dim7 is in the nuclear transport of proteins, our results surprisingly demonstrate that the role of Dim7 in embryonic myogenesis is independent of nuclear import; instead Dim7 is tethered to the muscle membrane at muscle attachment sites by integrin complexes and biochemically interacts with the Engulfment and Cell Motility (Elmo) protein to mediate actin cytoskeletal arrangement in actively contracting muscles.
Identify Mechanisms that Maintain Healthy Muscle in Ageing Organisms
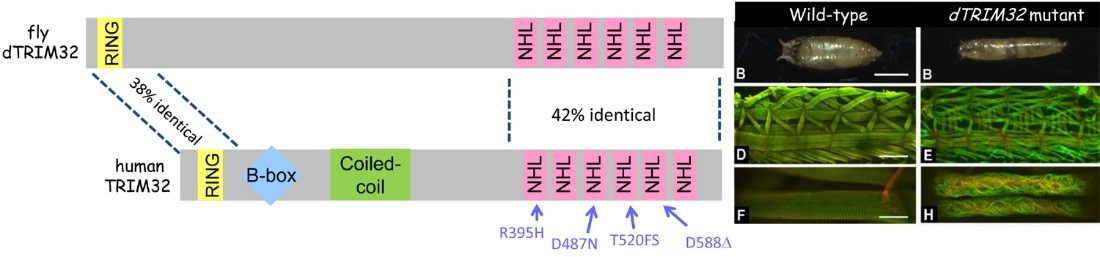
In addition to congenital muscular dystrophies, which are present at birth, muscle atrophy (or muscle wasting) is a problem in elderly adults or in patients who are compromised as a result of other illnesses, such as diabetes or cancer. We recently characterized a degenerative muscle phenotype in a mutant called thin (tn). tn mutants are pupal lethal and exhibit a thin, elongated pupal case consistent with the inability of the muscles to properly contract during pupariation. Thin is the single ortholog of vertebrate Trim32, a protein defective in patients affected with limb-girdle muscular dystrophy type 2H (LGMD2H). Our studies have revealed that Thin localizes to specific locations, called costameres, within mature muscles and is essential for both the integrity of the costamere and myofibril stability. Thin is a putative E3 ubiquitin ligase and may be important for protein stabilization (via monoubiquitination) or protein degradation (via polyubiquitination) within the muscle cell during normal homeostasis. Characterization of additional degenerative muscle mutants are in progress.
The Dock Family of Proteins in Morphogenesis of the Drosophila Heart Tube
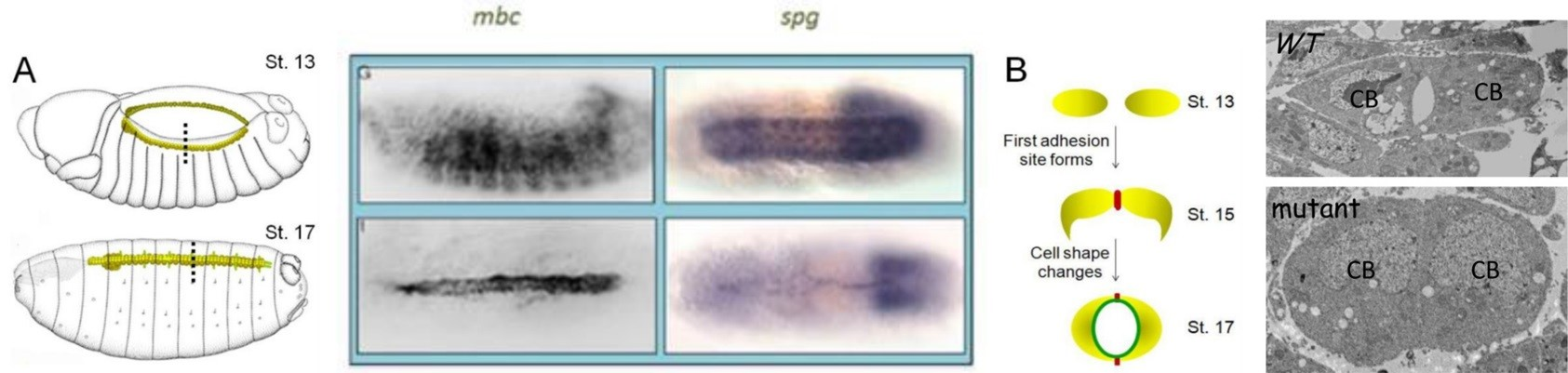
In our in vivo mass spectrometry approach to identify Elmo-interacting partners, we detected peptides corresponding to CG31048, also called Sponge (Spg). In a 2011 publication, we verified a physical interaction between Elmo and Spg and demonstrated a requirement for Spg downstream of N-cadherin in the developing central nervous system (CNS). Consistent with this, we found that Spg was highly expressed in the CNS and not detectable in muscle, where Mbc expression is high. Spg and Mbc share a similar domain structure and are orthologous to Dock4 and Dock180, respectively. While both Dock-Elmo complexes have been shown to activate Rac to modulate the cytoskeleton, Dock 4 is also reported to regulate cell adhesion via the GTPase Rap. The simplest hypothesis is that the Mbc-Elmo functions in muscle tissue and Spg-Elmo is required in the nervous system to activate Rac. However, expression of full length Spg in the muscle does not rescue mbc myoblast fusion defects, as observed with the expression of Mbc. These data suggest that the Dock family proteins Mbc and Spg are not functionally redundant in all cell or tissue-types. During our expression analysis, we observed high levels of both Mbc and Spg protein in the developing dorsal vessel. This simple heart tube is composed of two rows of cardioblasts, or heart cells, which change shape as they migrate towards one another to establish adhesion sites between the adjacent rows of cells. Such a simple morphogenic process results in the formation of a central lumen that is required for hemolymph circulation in the organism. Cell migration and cell adhesion are two cellular processes that contribute this process. Using the dorsal vessel as a model, our goal is to decipher the role of the Drosophila Dock-Elmo complexes in cell migration and cell adhesive events.